




合作客戶/
拜耳公司 |
同濟大學 |
聯合大學 |
美國保潔 |
美國強生 |
瑞士羅氏 |






相關新聞Info
含氟杯芳烴雙咪唑季銨鹽化合物1形成的LB膜為H-聚集體
來源:湖北師范大學學報(自然科學版) 瀏覽 165 次 發布時間:2024-07-16
合成了含氟杯芳烴離子鹽化合物1,利用表面壓一面積(π-A)等溫線、壓縮/擴張循環等溫線與膜穩曲線、紫外與紅外光譜等手段研究了該化合物在空氣/水相界面、空氣/二元羧酸水溶液界面的成膜性能及陰離子識別。結果表明:化合物1在水或者二元羧酸水溶液的亞相表面均能很好地形成穩定的Langmuir膜,在界面能識別二元羧酸陰離子,并能轉移到固體基片上形成LB膜,形成的LB膜為H-聚集體。
杯芳烴是繼冠醚和環糊精之后的第三代超分子主體化合物,其在客體分子識別方面,如中性分子、金屬離子、生物小分子等,有著非常好的應用前景,在超分子化學領域有著重要而廣泛的應用。Joseph等發現一種含有亞胺基杯芳烴受體對Zn2+有高選擇性識別,Chung等研究了β-氨基-α,β-不飽和酮取代的杯芳烴衍生物和異噁唑取代的杯芳烴衍生物對Cu2+具有選擇性識別,分別表現為熒光強度的增強和減弱。不僅如此杯芳烴也能識別陰離子,Liu等在杯芳烴的下緣導入酰胺和硫脲組合基團的化合物對直鏈α,β-二元羧酸具有高度的選擇性識別,當客體分子為己二酸雙陰離子時,由于氫鍵作用導致光誘導電子轉移,從而引起熒光強度的明顯變化,肉眼也能直接觀察到溶液的顏色變化,為有機陰離子的熒光檢測和比色分析提供了有效簡潔的方法。Singh等研究發現下沿修飾了氨基苯并咪唑的杯芳烴對庚二酸鹽具有選擇性識別,Stoikov等研究了一系列杯芳烴衍生物對多種二元羧酸和R-羥基羧酸的膜轉移性能。LB膜技術是一種精確控制薄膜厚度和分子排列的單分子膜沉積技術,即在水氣界面上能將成膜材料分子加以緊密有序的排列,形成單分子層,然后再轉移到固體襯底上的制膜技術,利用該技術可以將功能化合物的光學、電磁學、分子識別與催化等性質轉移到膜材料中。由于杯芳烴具有較好的建筑切塊功能,近年來利用杯芳烴構建的兩親分子Langmuir-Blodgett膜(LB膜)性質研究也引起了人們的極大興趣。最近我們合成了含氟杯芳烴離子鹽化合物1(圖1)并對該化合物在空氣/水相界面的成膜性能、二元羧酸陰離子識別等性質進行了研究,本文中報道相關的研究結果。
1實驗部分
1.1藥品和儀器
含氟杯芳烴離子鹽化合物1按參考文獻方法合成,其它所用試劑均為分析純,從國藥集團化學試劑有限公司購買,鍍膜用氯仿重蒸后使用,實驗用水為二次石英亞沸蒸餾水。π-A等溫曲線測量和LB膜制備在KSV-5000儀器上(芬蘭)進行,紫外光譜用U-3010紫外可見分光光度計(日本Hitachi公司)測定,紅外光譜用Niconet 5700光譜儀測定,檢測分辨率為4 cm-1,掃描32次消除基底噪音的影響。
1.2π-A等溫線測定和LB膜制備
將成膜材料化合物1溶解在重蒸的氯仿中,配成濃度為2×10-4mol/L的溶液,用微量注射器將一定量的溶液鋪展在LB膜分析儀槽內的亞相表面,亞相分別為二次蒸餾水或者含l.0mmol/L二元有機酸的純水,待氯仿溶劑靜止揮發20min后,在(20±0.5)℃的溫度下作表面壓-分子面積等溫線,壓膜速度為10 mm/min.在保持恒定的目標膜壓下,用水平轉移法將所得的多層膜轉移到石英基片上進行紫外光譜的測量。
壓縮/擴展等溫循環實驗:當膜壓達到預先設置的靶壓,停留5min后滑障反方向等速擴展至初始位置,在初始位置停留5min后,再反方向等速壓縮至設置的靶壓,來回循環3次,壓膜速度10mm/min.
膜穩定性實驗:將鋪展好的單分子膜壓縮至預先設置的表面壓(膜崩潰之前的壓力),然后停止壓膜,觀測化合物表面壓隨時間的變化。
2結果與討論
2.1表面壓一面積(π-A)等溫線
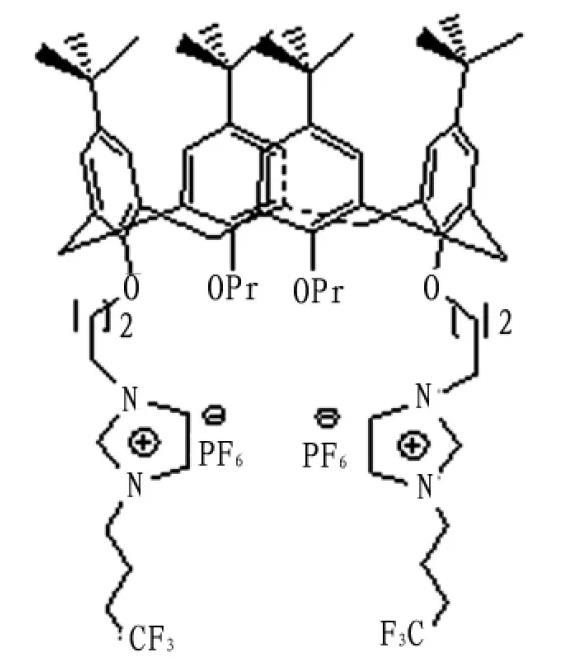
圖1杯芳烴雙咪唑季銨鹽化合物1的結構式
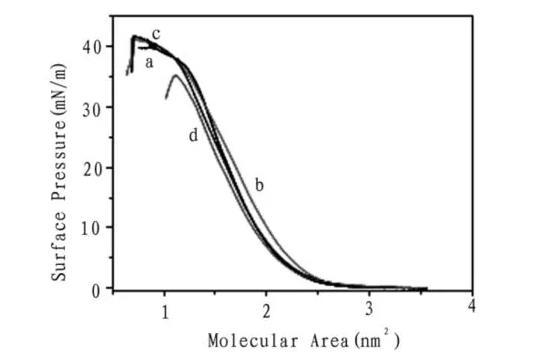
圖2化合物1在不同亞相π-A等溫線
圖2為化合物1在純水和濃度為1×10-3mol/L的乙二酸,丙二酸,丁二酸亞相上的表面壓-分子面積等溫線。結果表明化合物1在純水和二元有機羧酸亞相均能很好的形成Langmuir膜,從π-A曲線觀察到化合物1在純水亞相時的崩潰壓為38 mN/m,在乙二酸,丙二酸,丁二酸水溶液亞相上的崩潰壓分別為41 mN/m,43 mN/m,35 mN/m.作固相段曲線的切線,外推至表面壓為零時,可求得化合物1在純水和二元有機羧酸亞相的平均單分子面積分別為1.9 nm2,2.4 nm2,2.2 nm2和2.1 nm2.崩潰壓的增加和單分子面積變大均表明化合物1與二元有機羧酸存在相互作用,形成了穩定的Langmuir膜。
2.2壓縮/擴張循環等溫線與膜穩曲線
圖3給出了化合物1的單分子膜在崩潰壓之前20mN/m表面壓下的壓縮/擴張循環等溫線。三次壓縮/擴張循環之間表現出一定的差異,第二次壓縮與擴張循環比第一次壓縮/擴張循環表現出較大的滯后現象,第二次與第三次的壓縮/擴張循環基本接近,表明化合物1的壓縮-擴展性能表現的不穩定。化合物1的Langmuir膜穩定性也可以從圖3的膜穩曲線證明,在純水亞相上將Langmuir膜以10mm/min的速率壓縮至表面壓20mN/m,停止壓膜維持90min后其表面壓發生了先降低后增加的較小變化,在乙二酸,丙二酸,丁二酸的亞相上其表面壓隨時間的增加而逐漸有所減小,主要表現在前20min表面壓的變化較大,在所測的50min范圍內變化率分別為15%,18%,10%,然后均處于穩定狀態,表明化合物1和不同的二元有機羧酸在界面組裝形成的雜化Langmuir膜在恒定的壓力下隨著時間的變化有一個構象與組裝方式發生微調的過程,這可能與其分子結構中具有柔性的烷基鏈結構和含氟基團有關。
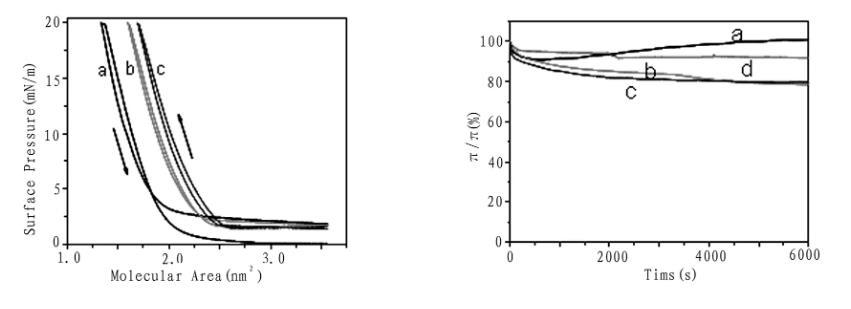
圖3化合物1的壓縮/擴張循環等溫線(左圖,a,b,c分別為1,2,3次)和不同亞相上的膜穩曲線(右圖,a純水,b乙二酸,c丙二酸,d丁二酸)
2.3紫外與紅外光譜
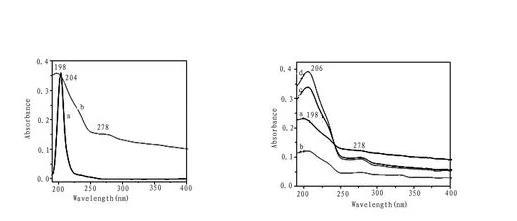
圖4化合物1氯仿溶液(a)和LB膜(30層)的紫外光譜(b)(左圖),在不同亞相(a)純水,(b)乙二酸,(c)丙二酸,(d)丁二酸時轉移30層膜的紫外光譜(右圖)
紫外吸收光譜常用來表征LB膜。圖4為化合物1在氯仿溶液和不同亞相時LB膜的紫外吸收光譜,可以看出溶液中204 nm處強吸收峰歸屬于化合物1中苯環的E2吸收帶,是由芳環的π→π*躍遷引起,轉移的LB膜吸收峰出現在198nm處,與溶液中吸收相比發生了6 nm的藍移,表明化合物1的LB膜組裝形成了H-聚集體。在乙二酸,丙二酸,丁二酸的水溶液三種二元羧酸陰離子亞相時的雜化膜的最大吸收分別在206nm處或者198nm處,并且在278nm處有一弱吸收,可能是由于杯芳烴下沿的咪唑環與二元羧酸陰離子發生靜電結合作用而使共軛增加,能量降低,從而吸收峰發生紅移的緣故。
圖5為化合物1在不同亞相時轉移30層膜的FT-IR圖,四種不同體系均在2958,2925和2861cm-1左右出現吸收峰,分別為杯芳烴兩親分子中芳環和脂肪鏈中C-H鍵的不對稱伸縮振動。純水亞相轉移的膜,在1552 cm-1和1465 cm-1出現芳環骨架振動吸收峰,亞相為乙二酸,丙二酸,丁二酸等二元羧酸時,其轉移的膜在1733 cm-1和1645 cm-1左右出現了有機羧基的特征伸縮振動,表明亞相中的二元羧酸陰離子與杯芳烴離子化合物1產生了離子識別,并通過靜電作用結合在一起轉移至基片上,得到杯芳烴雙陽離子與二元羧酸陰離子的雜化多層膜。
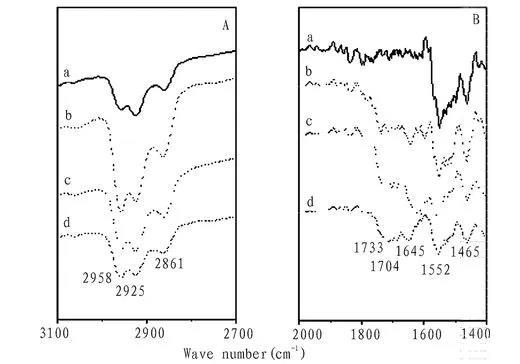
圖5化合物1在不同亞相時轉移30層膜的FT-IR圖譜
2.4離子識別機理
同樣是二元羧酸作為亞相,π-A等溫線會有如此大的差異,分析認為此差異可能歸結為化合物1與二元羧酸存在不同的結合方式。杯芳烴化合物在氣/液界面進行鋪展時,苯環形成的空腔和四個叔丁基作為疏水端朝向空氣,醚氧基和咪唑環作為親水端朝向水面,形成穩定的單分子固相膜。在亞相中加入乙二酸,丙二酸和丁二酸三種二元羧酸后,二元羧酸會電離生產羧酸根陰離子和氫陽離子,杯芳烴親水端的咪唑正離子部分可以與二元羧酸陰離子部分通過靜電作用力進行結合。而亞相的二酸陰離子的碳鏈長度不同,使得其與杯芳烴分子發生作用的方式也不同。通過CPK模型計算可得,乙二酸,丙二酸和丁二酸的長度分別為0.34nm,0.39nm,0.61nm,而化合物1的杯芳烴分子中雙咪唑環的間隔長度為0.73nm,這樣結合方式就有分子間和分子內兩種方式。當亞相為乙二酸和丙二酸時,由于雙咪唑環距離較遠,乙二酸和丙二酸分子較短,乙二酸和丙二酸分子只有置于兩個杯芳烴分子咪唑環的中間才能使得單層膜較穩定(如圖6),使得平均單分子面積增大,此為分子間排列。亞相為丁二酸時,丁二酸的長度與杯芳烴分子雙咪唑環的間距相差不多,丁二酸置于單個杯芳烴分子雙咪唑環的內部可以更加穩定,使得雙咪唑環略微收縮,平均單分子面積與純水亞相時持平或略有減少,此為分子內排列。
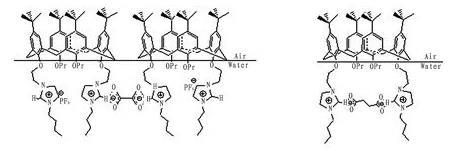
圖6化合物1與乙二酸和丁二酸在界面存在的分子間和分子內結合方式
3結論
合成了含氟杯芳烴雙咪唑季銨鹽化合物1,并對該化合物在空氣/水相界面的成膜性能以及對二元羧酸陰離子識別性質進行了研究,利用表面壓一面積(π-A)等溫線、壓縮/擴張循環等溫線與膜穩曲線、紫外與紅外光譜等手段表明化合物1在水或者二元羧酸水溶液的亞相表面均能很好的形成穩定的Langmuir膜,并能與二元羧酸陰離子一起轉移到固體基片上形成LB膜,化合物1形成的LB膜為H-聚集體。